
2019版原发性肉碱缺乏症筛查与诊治共识(全文)
2025-08-26
999+
74.5KB
12 页
海报
侵权投诉
2019 版:原发性肉碱缺乏症筛查与诊治共识(全文)
原发性肉碱缺乏症(PCD)又称原发性肉碱吸收障碍(CUD),或肉
碱转运障碍(CTD),是由于 SLC22A5 基因突变引起高亲和力钠依赖性
肉碱转运体(OCTN2)蛋白功能缺陷,尿中肉碱排出增加,血液、组织、
细胞内肉碱缺乏,从而引起脂肪酸 β 氧化缺陷的疾病。PCD 患病率具有
明显种族差异。美国报道患病率为 1/20 000~70 000[1]、日本为 1/40
000[2]、澳大利亚为 1/120 000[3],该病在法罗群岛患病率最高,为
1/300[4] , 中 国 报 道 的 新 生 儿 筛 查 PCD 患 病 率 约 为 1/20 000~45
000[5,6]。我国上海新华医院报道发病率为 1/34 571,浙江省新筛中心
对近 213 万新生儿筛查,确诊 101 例 PCD 患儿,患病率为 1/21 089。
由于 PCD 临床表现具有异质性及非特异性,易误诊或漏诊,部分患者可
终生无异常表现。发病的患者未经治疗具潜在致死性。此病药物治疗效
果确切,在脏器功能发生不可逆损伤前补充左卡尼汀治疗者预后良好。
早期诊断、早期治疗可明显改善预后,故许多国家及地区将其列入新生
儿筛查病种[7]。
随着我国新生儿遗传代谢病筛查的广泛开展,串联质谱技术应用于新
生儿遗传病筛查得到进一步推广,更多的 PCD 患儿得到检出,但目前尚
无统一的本病筛查、诊治以及随访共识。为了规范 PCD 新生儿筛查的流
程及后续的诊断和遗传咨询,由中华预防医学会出生缺陷预防与控制专
业委员会新生儿遗传代谢病筛查学组、中华医学会儿科分会出生缺陷预
防与控制专业委员会、中国医师协会医学遗传医师分会临床生化遗传专
业委员会、青春期医学专业委员会临床遗传学组、中华医学会儿科分会
临床营养学组及《中华医学杂志》编辑委员会组织专家讨论,并达成以
下共识。
一、病因及发病机制
肉碱是一种类氨基酸物质,化学名为 3-羟基-4-氮-三甲氨基丁酸,分
为左旋肉碱及 D-肉碱,左旋肉碱具有生理活性。体内肉碱约 75%来源胃
肠道食品摄入 ( 主 要 是瘦肉食 品 ),约 25% 来源 于体内自身合成
[8]。PCD 是由于 SLC22A5 基因突变致细胞膜上 OCTN2 功能缺陷的常
染色体隐性遗传病。OCTN2 存在于肠黏膜、肝脏、心肌、骨骼肌及肾小
管等组织细胞膜上,将肉碱由细胞外转运至细胞内。肠道细胞 OCTN2 功
能缺陷导致肉碱通过胃肠道进入血液受阻,肾脏疾病(肾功能不全)所
致的 OCTN2 功能缺陷可使肾小管重吸收肉碱障碍,尿肉碱排泄增加,这
2 种因素均可导致血浆肉碱水平降低。各脏器(主要是肝脏、心肌及骨骼
肌)OCTN2 功能缺陷致组织细胞内肉碱进一步缺乏,引起细胞功能障碍。
肉碱的主要功能是将中、长链脂肪酸从细胞质转运至线粒体内进行脂
肪酸 β 氧化的必须载体。脂肪酸 β 氧化是为肝脏、心肌、骨骼肌提供能
量的主要形式,肉碱缺乏致脂肪酸 β 氧化受阻,可造成低血糖及酮体减
少,组织细胞内能量供应不足,导致细胞损伤,肝酶及肌酸激酶增高。
脂肪利用减少,积聚在肝脏、骨骼肌、心肌,导致肝细胞脂肪变性和肌
病[8,9]。
编码 OCTN2 的 SLC22A5 基因位于 5q31.1,含10 个外显子,约 3.2
kb 长。OCTN2 是一种跨膜蛋白,由 557 个氨基酸组成,包含 12 个跨膜

位点及 ATP 结合位点。已报道的致病性突变超过 180 种(APUP 实验室
SLC22A5 数据库,http://www.arup.utah.edu/database/OCTN2/
OCTN2_display.php),约一半致病性突变为错义突变,其余的为无义
突变、剪 接 突变、小片段插入或缺失。HGMD数据库
(www.hgmd.cf.ac.uk)还收录了 6 种大片段缺失[7]。我国常见突变为
c.760C>T(p. R254X),约占25.6%[10]。
二、新生儿筛查
依据PCD 在我国具有较高的患病率,并且左卡尼汀治疗本病效果显
著,故此病在我国值得进行新生儿筛查,以便达到早发现、早治疗的目
的。
1.PCD 新生儿筛查方法:
目前只有串联质谱(MS/MS)能够对血液中游离肉碱(C0)及不同
种类酰基肉碱进行快速、特异及准确地检测,故依据我国《新生儿疾病
筛查管理办法》及《新生儿疾病筛查技术规范》,可利用串联质谱对新
生儿进行PCD 筛查。新生儿出生后 48 h 采血,滴于专用滤纸片上,晾干,
送检,新生儿筛查中心及时进行血液 C0 及各种酰基肉碱检测。
2.游离肉碱检测切值:
新生儿期与非新生儿期干血滤纸片 C0 参考范围略有不同;样品处理
衍生法与非衍生法略有不同;不同实验室之间略有不同。新生儿及非新
生儿 C0 低限均为 10 μmol/L;新生儿 C0 上限为 50~60 μmol/L,非新
生儿 C0 上限为 60~90 μmol/L(样品处理方法、不同实验室之间有差
异)。

摘要:
展开>>
收起<<
2019版:原发性肉碱缺乏症筛查与诊治共识(全文)原发性肉碱缺乏症(PCD)又称原发性肉碱吸收障碍(CUD),或肉碱转运障碍(CTD),是由于SLC22A5基因突变引起高亲和力钠依赖性肉碱转运体(OCTN2)蛋白功能缺陷,尿中肉碱排出增加,血液、组织、细胞内肉碱缺乏,从而引起脂肪酸β氧化缺陷的疾病。PCD患病率具有明显种族差异。美国报道患病率为1/20000~70000[1]、日本为1/40000[2]、澳大利亚为1/120000[3],该病在法罗群岛患病率最高,为1/300[4],中国报道的新生儿筛查PCD患病率约为1/20000~45000[5,6]。我国上海新华医院报道发病率为1/345...
相关推荐
-
妇科诊疗技术操作标准规范_20622084

 2025-11-29 999+
2025-11-29 999+ -
妇科诊疗常规(已审)_20625126

 2025-11-29 999+
2025-11-29 999+ -
妇科人流综合症应急预案及流程_20622076
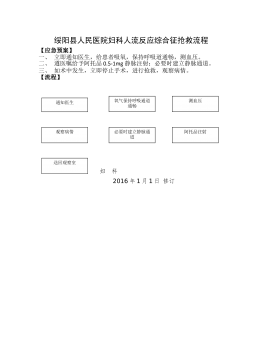 2025-11-29 999+
2025-11-29 999+ -
妇科检查及二合诊三合诊图解_20622068
 2025-11-29 999+
2025-11-29 999+ -
妇科检查规范_20622067
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规讲解_20625121
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规_20622061
 2025-11-29 999+
2025-11-29 999+ -
妇科 不孕病(多囊卵巢综合征)中医临床路径_20625117
 2025-11-29 999+
2025-11-29 999+ -
二级病原微生物试验室生物安全管理规范-上海卫生和计划生育_20621748
 2025-11-29 999+
2025-11-29 999+ -
2022子宫内膜异位症【29页】

 2025-11-29 999+
2025-11-29 999+

