
运动神经元病临床诊断治疗指南
2025-08-24
999+
30KB
18 页
海报
侵权投诉
运动神经元病临床诊断治疗指南
【概述】
运动神经元病(motor netlron disease,MND)为一组病因
不清的系统性疾病。主要累及上、下运动神经元的慢性进
行性变性疾病。根据受损的病变部位不同而分为进行性脊
肌萎缩症、进行性延髓麻痹、原发性侧索硬化和肌萎缩侧
索硬化数种类型。
一、肌萎缩侧索硬化
【临床表现】
肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)
可分为三型,即散发型(或称经典型)、家族型(5%~10%)
和关岛型。
散发型表现为中年或中年以后起病,40 岁以下起病者亦不
少见。国内报告最早发病为 18 岁。男性多于女性。起病隐
袭,呈慢性进展病程,部分患者为亚急性病程,少数起病
后呈急剧进展,可于病后半年左右死亡。早期出现肌肉无
力、肌肉萎缩及肌纤颤。常自上肢远端手部肌肉开始,可
自一侧手肌开始,数月后可波及对侧;也可双侧手肌同时
受累,随后波及前臂肌肉及上臂和肩胛部肌肉。部分患者
可以三角肌或冈上、下肌无力开始,造成肩胛下垂,抬肩
或臂上举无力。少数患者可以下肢起病,表现为下肢无力、
沉重、走路无力,骨盆带肌肉受累后可有上台阶、楼梯、
蹲下起立困难,下肢肌肉萎缩。随着病情的发展,肌无力
和萎缩可延至颈部、躯干、面肌及延髓支配的肌肉,表现
为抬头困难、转颈障碍,呼吸肌受累出现呼吸困难,延髓
支配肌肉受累则有吞咽困难、咀嚼费力、舌肌萎缩、发音
障碍等。延髓麻痹通常出现于疾病晚期,但也可于手肌萎
缩不久后出现,少数情况下为首发症状。肌纤颤为常见的
症状,可在多个肢体中发生。在舌体由于肌膜薄而可看到
肌纤维颤动。本病很少有感觉障碍,客观感觉异常少见,
有少数患者可有痛性痉挛。绝大多数患者无括约肌障碍。
本病的生存期随临床类型而有不同,短者数月,长者可十
余年,最长可至 35 年,平均生存 3年左右。严重者多因延
髓麻痹或呼吸麻痹而死亡。
本病患者在肌肉萎缩的同时出现上运动神经元损害体征,
表现为腱反射亢进,手部可引出Hoffmann 及 Rossolimo 等
病理征。在以上肢起病的患者,下肢可出现肌肉痉挛,肌
张力增高,反射亢进,Babinski征及Chaddock 征阳性。在
皮质延髓束受累的情况下,可出现下颌反射亢进及强哭强
笑等假性延髓麻痹
【诊断要点】
(一)诊断
经临床、电生理、影像学证实有上运动神经元和下运动神
经元病损的患者,在排除有相似临床表现的疾病,特别是
可治疗的疾病如脊髓压迫症等后,可以作出临床诊断。
(二)实验室检查
一般实验室检查,旨在排除诊断之用。
1.脑脊液检查多正常,少数可有蛋白轻度增高。
2.血液生化血肌酶谱多为正常,在进展的疾病中可有增高。
尿中肌酸 HJ 轻度增高,肌酐排出减少。
3.免疫学检查血中免疫球蛋白及补体在正常范围。血清中
球蛋白增高司见于少数患者,推测与组织及细胞坏死有关。
(三)电生理检查
1.肌电图 肌电图检查能提示肌萎缩为神经源性,为下运
动神经元受损提供电生理证据。其表现为静止状态示插入
电位延长,出现纤颤电位,正锐波。轻收缩状态运动单位
电位(MUAP)时限延长,波幅增高,多相电位增多。重收缩
状态由于运动单位数量减少,不出现干扰相,而为单纯相。
在慢性进展、病程长、芽生能力强时可出现巨大电位,但
并非前角细胞病变所特有。值得提出的是,在肌电图检查
时选择肌肉应避免极度萎缩的肌肉,因为在此种情况下阳
性率会受到影响。胸锁乳突肌肌电图的阳性率达 97%。

摘要:
展开>>
收起<<
运动神经元病临床诊断治疗指南【概述】运动神经元病(motornetlrondisease,MND)为一组病因不清的系统性疾病。主要累及上、下运动神经元的慢性进行性变性疾病。根据受损的病变部位不同而分为进行性脊肌萎缩症、进行性延髓麻痹、原发性侧索硬化和肌萎缩侧索硬化数种类型。一、肌萎缩侧索硬化【临床表现】肌萎缩侧索硬化(amyotrophiclateralsclerosis,ALS)可分为三型,即散发型(或称经典型)、家族型(5%~10%)和关岛型。散发型表现为中年或中年以后起病,40岁以下起病者亦不少见。国内报告最早发病为18岁。男性多于女性。起病隐袭,呈慢性进展病程,部分患者为亚急性病程,少...
相关推荐
-
妇科诊疗技术操作标准规范_20622084

 2025-11-29 999+
2025-11-29 999+ -
妇科诊疗常规(已审)_20625126

 2025-11-29 999+
2025-11-29 999+ -
妇科人流综合症应急预案及流程_20622076
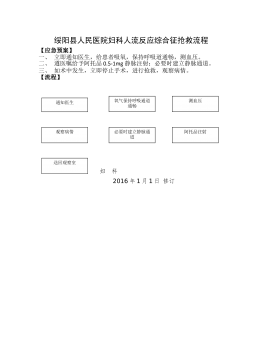 2025-11-29 999+
2025-11-29 999+ -
妇科检查及二合诊三合诊图解_20622068
 2025-11-29 999+
2025-11-29 999+ -
妇科检查规范_20622067
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规讲解_20625121
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规_20622061
 2025-11-29 999+
2025-11-29 999+ -
妇科 不孕病(多囊卵巢综合征)中医临床路径_20625117
 2025-11-29 999+
2025-11-29 999+ -
二级病原微生物试验室生物安全管理规范-上海卫生和计划生育_20621748
 2025-11-29 999+
2025-11-29 999+ -
2022子宫内膜异位症【29页】

 2025-11-29 999+
2025-11-29 999+

