
《脊髓性肌萎缩症多学科管理专家共识》(2019)要点
2025-08-22
999+
18.5KB
3 页
海报
侵权投诉

《脊髓性肌萎缩症多学科管理专家共识》(2019)要点
脊髓性肌萎缩症(SMA)是由于脊髓前角及延髓运动神经元
变性,导致近端肢体和躯干进行性、对称性肌无力和肌萎缩的神
经变性病。尽管 SMA 可由多种基因突变引起,但一般特指由于运
动神经元存活基因 1 (SMNl)突变所导致的常染色体隐性遗传病。
目前中国尚无发病率的确切数据,中国人群中的携带者频率约为
1/42。随着病情的进展,肌无力可进一步导致骨骼系统、呼吸系
统、消化系统及其他系统异常,其中呼吸衰竭是最常见的死亡原
因。因此,SMA 的治疗往往需要多学科的评估和综合管理。
一、临床表现
该病临床表现差异较大,根据患者起病年龄和所获得的最大
运动功能,将 SMA 由重到轻分为 4 型。
1.1 型:也称 werdnig-Hoffman 病,即婴儿型,约占全
部 SMA 病例的 45%。患儿出生后 6 个月内起病,出现迅速发展的
进行性、对称性四肢无力,最大运动能力不能达到独坐。肌无力
以近端为著,由于显著肌张力低下,平躺时下肢呈“蛙腿”样姿
势。
2.2 型 :也称 Dubowitz 病 , 即中间型,约占 30 % ~
40%。患者多在生后 6~18 个月起病,进展较 1 型慢,最大运动能
力可达到独坐,但独坐年龄可能落后于正常同龄儿,不能独站或
独走。
3.3 型:也称 Kugelberg-Welander 病,即青少年型,
约占 20%。患者多在出生 18 个月后起病,早期运动发育正常,可
独走,部分独走时间延迟。随年龄增长出现以近端为主的肌无力,
下肢重于上肢,最终部分丧失独走能力,逐渐依赖轮椅。
4.4 型:晚发型,即成人型,早期运动发育正常,成人
起病,出现肢体近端无力,进展缓慢,预期寿命不缩短。
二、发病机制
位于染色体 5q11.2-q13.3 的 SMNl 基因是 5qSMA 的主要致病
基因,SMNl 基因致病性突变引起编码的运动神经元存活蛋白表达
水平下降或功能丧失。
三、检查项目与诊断流程
1.检查项目:临床疑诊为 SMA 的患者,即表现为躯干
和四肢近端肢体为主的进行性、对称性肌无力和肌萎缩、肌束颤 、
腱反射减弱或消失、智力正常,可以选择以下辅助检查以明确诊
断。
(1)基因检测:
(2)血清肌酸激酶(CK):
(3)肌电图:
(4)肌肉病理:

摘要:
展开>>
收起<<
《脊髓性肌萎缩症多学科管理专家共识》(2019)要点如下:脊髓性肌萎缩症(SMA)是神经变性病主要由SMN1基因突变导致中国人群携带者频率约为142。其临床表现差异大根据起病年龄和最大运动功能分为4型病情严重程度依次递减。该病由SMN1基因致病性突变引起检查项目包括基因检测、血清肌酸激酶、肌电图和肌肉病理等。其中基因检测是确诊SMA的关键手段。由于SMA涉及多系统损害多学科综合管理至关重要包括多专业评估、康复治疗、骨科脊柱外科管理、消化系统和营养管理、呼吸功能管理、紧急医疗救护、免疫接种、心理治疗、病人转诊与平稳过渡以及遗传咨询和产前诊断。其中康复治疗是干预、延缓疾病进展的主要手段。此共识基于现有证据制定未来会不断更新。
相关推荐
-
妇科诊疗技术操作标准规范_20622084

 2025-11-29 999+
2025-11-29 999+ -
妇科诊疗常规(已审)_20625126

 2025-11-29 999+
2025-11-29 999+ -
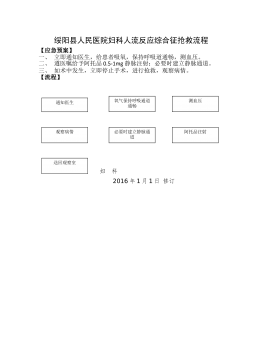
妇科人流综合症应急预案及流程_20622076
 2025-11-29 999+
2025-11-29 999+ -
妇科检查及二合诊三合诊图解_20622068
 2025-11-29 999+
2025-11-29 999+ -
妇科检查规范_20622067
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规讲解_20625121
 2025-11-29 999+
2025-11-29 999+ -
妇科护理常规_20622061
 2025-11-29 999+
2025-11-29 999+ -
妇科 不孕病(多囊卵巢综合征)中医临床路径_20625117
 2025-11-29 999+
2025-11-29 999+ -
二级病原微生物试验室生物安全管理规范-上海卫生和计划生育_20621748
 2025-11-29 999+
2025-11-29 999+ -
2022子宫内膜异位症【29页】

 2025-11-29 999+
2025-11-29 999+

